Probably is the reaction of ordinary steel with its surroundings, where a voluminous, and porous coat of rust is formed, the most well-known example of what is called corrosion. There are many acquaintances examples from everyday life such as the rusting of garden trellises, steel structures, ships the rusting of bolts and nuts or exhausts of cars etc. Often, corrosion and rust are synonym.
One general definition of corrosion is the degradation of a material through environmental interaction. This definition encompasses all materials, both naturally occurring and man-made and includes plastics, ceramics, and metals. This Introduction focuses on the corrosion of metals, with emphasis on corrosion of carbon and low-alloy steel used in constructions, ships, bridges etc. This definition of corrosion begs the question; why do metals corrode? The answer lies in the field of thermodynamics, which tells whether a process such as corrosion will occur. A second logical question is what is the rate of corrosion or how long will, for instance, a pipeline last? Corrosion kinetics can help provide an answer to this question. Both topics are discussed in greater detail in a future paper called “Fundamentals of Corrosion”.
A significant amount of energy is put into a metal when it is extracted from its ores, placing it in a high-energy state. These ores are typically oxides of the metal such as hematite (Fe2O3) for steel or bauxite (Al2O3¢H2O) for aluminium. One principle of thermodynamics is that a material always seeks the lowest energy state. In other words, most metals are thermodynamically unstable and will tend to seek a lower energy state, which is an oxide or some other compound. The process by which metals convert to the lower-energy oxides is called corrosion.
Corrosion of most common engineering materials at near-ambient temperatures occurs in aqueous (water-containing) environments and is electrochemical in nature. The aqueous environment is also referred to as the electrolyte and, in the case of underground corrosion, is moist soil. The corrosion process involves the removal of electrons (oxidation) of the metal [Equation (1)] and the consumption of those electrons by some other reduction reaction, such as oxygen or water reduction [Equations (2) and (3),
respectively:
Fe → Fe++ + 2e- (1)
O2 + 2H2O + 4e- → 4OH- (2)
2H2O + 2e- + 2OH- (3)
The oxidation reaction is commonly called the anodic reaction and the reduction reaction is called the cathodic reaction. Both electrochemical reactions are necessary for corrosion to occur. The oxidation reaction causes the actual metal loss, but the reduction reaction must be present to consume the electrons liberated by the oxidation reaction, maintaining charge neutrality. Otherwise, a large negative charge would rapidly develop between the metal and the electrolyte and the corrosion process would cease.
The oxidation and reduction reactions are sometimes referred to as half-cell reactions and can occur locally (at the same site on the metal) or can be physically separated. When the electrochemical reactions are physically separated, the process is referred to as a differential corrosion cell. A schematic of a differential corrosion cell is given in Figure 1.1. The site where the metal is being oxidized is referred to as the anode or anodic site. At this site, direct electric current (defined as a positive flow of charge) flows from the metal surface into the electrolyte as the metal ions leave the surface. This current flows in the electrolyte to the site where oxygen, water, or some other species is being reduced. This site is referred to as the cathode or cathodic site. There are four necessary components of a differential corrosion cell.
1. There must be an anode
2. There must be a cathode
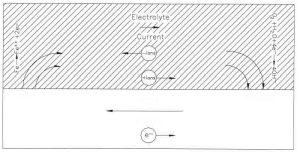
Figure 1.1 Schematic showing a differential corrosion cell
3. There must be a metallic path electrically connecting the anode and cathode. (Normally, this will be the construction itself.)
4. The anode and cathode must be immersed in an electrically conductive electrolyte (moist soil, water etc.).
Underground corrosion of pipelines and other structures is often the result of differential corrosion cells of which a variety of different types exist. These include differential aeration cells, where different parts of a pipe are exposed to different oxygen concentrations in the soil, and cells created by differences in the nature of the pipe surface or the soil chemistry. Galvanic corrosion is a form of differential cell corrosion in which two different metals are electrically coupled and exposed in a corrosive environment.
Further discussion of these differential corrosion cells is given below and in the paper “Fundamentals of Corrosion”.
Types of corrosion
There are several types of corrosion, each of which can be classified by the cause of the metal`s chemical deterioration.
Listed below are 10 common types of corrosion:
Localized Corrosion:
Unlike general attack corrosion, localized corrosion specifically targets one area of the metal structure. Localized corrosion is classified as one of three types:
- Pitting: Pitting results when a small hole, or cavity, forms in the metal, usually as a result of de-passivation of a small area. This area becomes anodic, while parts of the remaining metal becomes cathodic, producing a localized galvanic reaction. The deterioration of this small area penetrates the metal and can lead to failure. This form of corrosion is often difficult to detect due to the fact that it is usually relatively small and may be covered and hidden by corrosion-produced compounds.
- Crevice corrosion: Similar to pitting, crevice corrosion at a specific location. This type of corrosion is often associated with a stagnant micro-environment, like those found under gasket and washers and clamps. Acidic conditions or a depletion of oxygen in a crevice can lead to crevice corrosion.
- Filiform corrosion: Occurring under painted or plated surfaces when water breaches the coating, filiform corrosion begins at small defects in the coating and spreads to cause structural weakness.
Galvanic Corrosion:
Galvanic corrosion, or dissimilar metal corrosion, occurs when two different metals are located together in a corrosive electrolyte. A galvanic couple forms between two metals, where one metal becomes the anode and the other the cathode. The anode, or sacrificial metal, corrodes and deteriorates faster than it would alone, while the cathode deteriorates more slowly than it would otherwise.
Three conditions must exist for galvanic corrosion to occur:
- Electrochemically dissimilar metals must be present
- The metals must be in electrical contact, and
- The metals must be exposed to an electrolyte
Environmental Cracking:
Environmental cracking is a corrosion process that can result from a combination or environmental conditions affecting the metal. Chemical, temperature and stress related conditions can result in the following types of environmental corrosion:
- Stress Corrosion Cracking (SCC)
- Hydrogen-induced cracking
- Liquid metal embrittlement
Flow-Assisted Corrosion (FAC):
Flow-assisted corrosion, or flow-accelerated corrosion, results when a protective layer of oxide on a metal surface is dissolved or removed by wind or water, exposing the underlying metal to further corroding and deteriorate.
Erosion-assisted corrosion
- Impingement
- Cavitation
Intergranular corrosion:
Intergranular corrosion is a chemical or electrochemical attack on the grain boundaries of a metal. It often occurs due to impurities in the metal, which tend to be present in higher contents near grain boundaries. These boundaries can be more vulnerable to corrosion than the bulk of the metal.
De-Alloying:
De-alloying, or selective leaching, is the selective corrosion of a specific element in an alloy. The most common type of de-alloying is de-zincification of unstabilized brass. The result of corrosion in such cases is a deteriorated and porous copper.
Fretting corrosion:
Fretting corrosion occurs as a result of repeated wearing, weight and/or vibration on an uneven, rough surface. Corrosion, resulting in pits and grooves, occurs on the surface.
Fretting corrosion is often found in rotation and impact machinery, bolted assemblies and bearings, as well as to surfaces exposed to vibration during transportation.
High-Temperature Corrosion:
Fuels used in gas turbines, diesel engines and other machinery, which contain vanadium or sulphates can, during combustion, form compounds with a low melting point. These compounds are very corrosive towards metal alloy normally resistant to high temperatures and corrosion, including stainless steel.
High-temperature corrosion can also be caused by high-temperature oxidization, sulfidation, and carbonization.
How do we detect Corrosion of underground structures?
The electrochemical nature of the corrosion process provides opportunities to detect and mitigate corrosion of underground structures. We can monitor the voltages and the currents associated with the corrosion process. When a piece of metal is placed in an electrolyte, such as soil, a voltage will develop across the metal–electrolyte interface because of the electrochemical nature of the corrosion process. We cannot measure this voltage directly but, using a voltmeter, we can measure a voltage between two different metals that are placed in the soil. We also can measure the voltage difference between a metal and a reference electrode, commonly called a half-cell electrode. This voltage is referred to as a corrosion potential, an open circuit potential, or a native potential for that metal in the environment in which the measurement is being obtained. For soil environments, the most common reference electrode used is the copper–copper sulphate reference electrode (CSE). Potential measurements can be used to estimate the relative resistance of different metals to corrosion in a given environment. Noble metals, such as gold and platinum, have more positive potentials and are more resistant to corrosion than are the more common engineering metals such as steel and aluminium. A galvanic series is a list of metals and alloys arranged according to their relative corrosion potentials in a given environment. Table 1.1 shows a galvanic series for metals and other materials in neutral soils and water, indicating that carbon has the most positive potential of the materials listed and magnesium has the most negative potential. The potentials measured for the different metals in a galvanic series vary somewhat, depending on the nature of the environment, but the relative position of the metals is similar for natural environments such as soil and seawater.
Table 1.1 Practical Galvanic Series for Materials in Neutral Soils and Water
Material Potential Volts (CSE)a
Carbon, Graphite, Coke +0.3
Platinum 0 to -0.1
Mill Scale on Steel -0.2
High Silicon Cast Iron -0.2
Copper, Brass, Bronze -0.2
Mild Steel in Concrete -0.2
Lead -0.5
Cast Iron (Not Graphitized) -0.5
Mild Steel (Rusted) -0.2 to -0.5
Mild Steel (Clean and Shiny) -0.5 to -0.8
Commercially Pure Aluminium -0.8
Aluminium Alloy (5% Zinc) -1.05
Zinc -1.1
Magnesium Alloy (6% Al, 3% Zn, 0.15% Mn) -1.6
Commercially Pure Magnesium -1.75
A Typical potential normally observed in neutral soils and water, measured with respect to copper sulphate reference electrode.
Another use for corrosion potential measurements is to establish whether galvanic corrosion is likely to occur. When two metals are electrically coupled in an environment, the more negative (active) member of the couple will become the anode in the differential corrosion cell, and the more positive (noble) member of the couple will become the cathode in the cell in general, the severity of the galvanic couple increases as the difference in potential between the two members of the couple increases, although this is not always the case. The galvanic series shown in Table 1.1 indicates that, where copper is electrically coupled to mild steel in soil, the copper will become the cathode and the steel will become the anode, accelerating corrosion of the steel. A further discussion of galvanic corrosion is given in the paper called “Fundamentals of Corrosion”.
Table 1.1 also shows that the potential of mild steel can differ depending on whether the surface is clean or covered with mill scale. The potential of steel also is a function of soil properties, including pH, ion concentration, oxygen, and moisture content. The potential differences that develop on underground pipelines and other structures as a result of these factors can result in severe corrosion. Further discussions of these differential corrosion cells are given in the paper called “Fundamentals of Corrosion”.
Potential measurements are commonly used on underground pipelines to detect the presence of these types of differential corrosion cells. An electrical connection is made to the pipe, and the potential of the pipe is measured with respect to a reference electrode placed over the pipe. This process is shown schematically in Figure 1.2. Normally, the reference electrode is connected to the negative lead of a digital voltmeter to obtain a negative reading. As shown in Table 1.1, most potentials in soils are negative. With this type of measurement, the most negative regions of the structure are the anodes and are undergoing accelerated corrosion due to the differential corrosion cells.
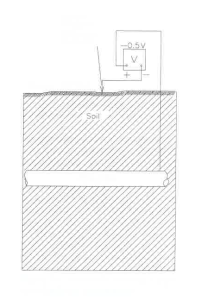
Figure 1.2 Schematic showing a pipe-to-soil potential measurement
Current measurements also can be used to detect differential corrosion cells if the anodes and cathodes are large. These large cells create long-line currents that can be detected by measurements made over the pipe or other underground structure. Through Ohm’s law (V = IR, where V is the voltage, I is the current, and R is the resistance) we know that current flow in the soil will create a voltage gradient. This gradient can be detected by placing identical reference electrodes over the pipe and measuring the voltage difference. The voltage measurements can be used to indicate the direction of the differential cell current. The anodic and cathodic sites on the pipeline can be located by performing a series of cell-to-cell potential measurements taken along the pipeline. Another possible source of current flow in the ground is stray currents.